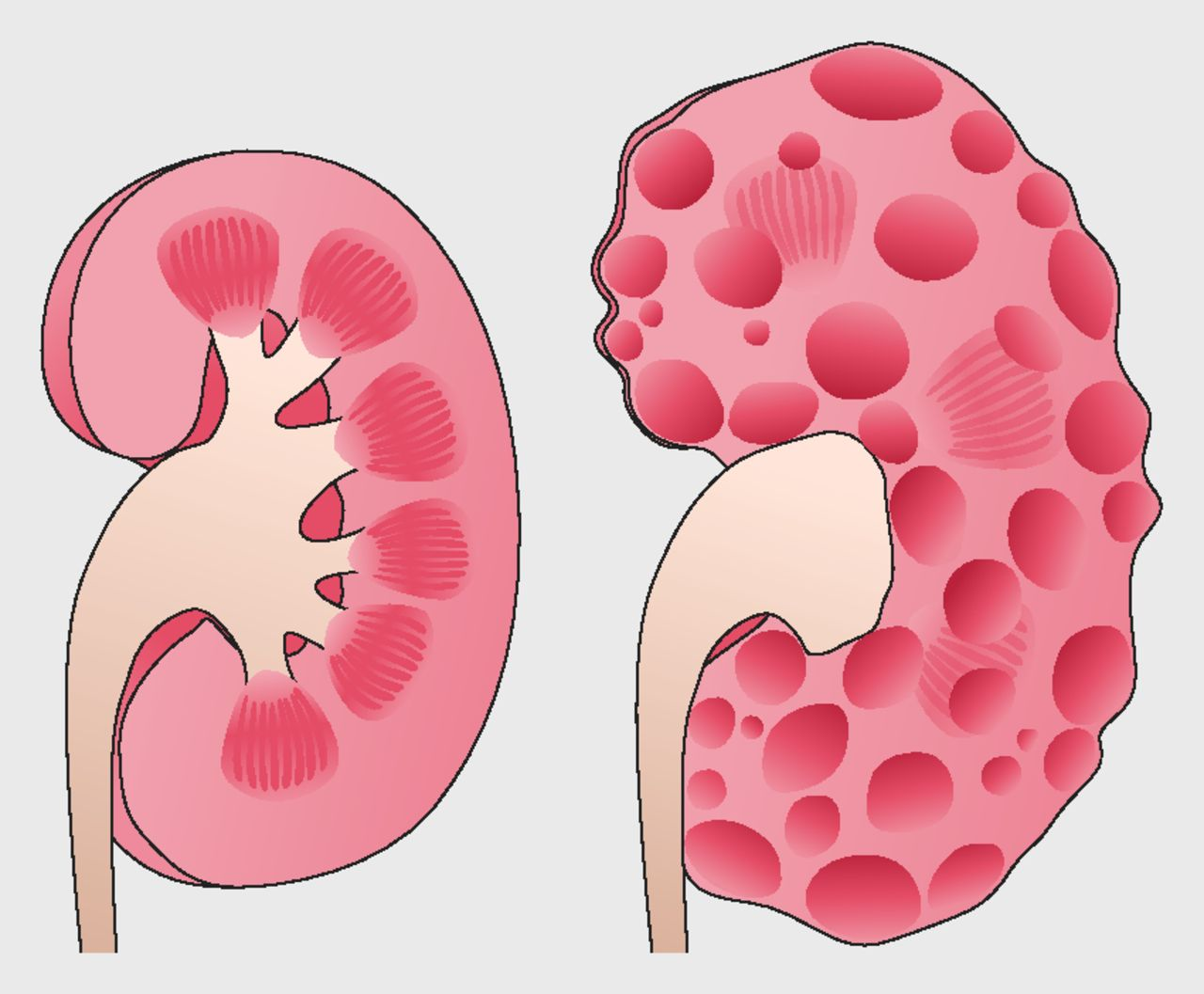
提到多囊肾,很多人的第一反应可能是"听起来很可怕"。
确实,这个名字听起来就让人联想到肾脏里面长了很多囊肿,像是肾脏变成了蜂窝煤一样。
但其实,多囊肾虽然是一种严重的遗传性疾病,但只要了解它的本质和发展规律,我们就能更好地应对它。
多囊肾病,医学上称为PKD,是一种由基因突变引起的遗传性肾脏疾病。
说得通俗一点,就是父母把"坏"基因传给了孩子,导致孩子的肾脏里面不断长出液体填充的囊肿。
这些囊肿会越长越大、越长越多,最终压迫正常的肾脏组织,影响肾脏的正常功能。
根据最新的医学统计数据,多囊肾病是人类最常见的遗传性肾脏疾病之一,全球发病率大约在1/400到1/1000之间。
更令人担忧的是,它是导致终末期肾病的第四大原因,仅次于慢性肾小球肾炎、糖尿病肾病和良性肾小动脉硬化。
在需要透析治疗的患者中,约有5%是由多囊肾病引起的。
这个疾病的特点是进展缓慢但不可逆转。
大多数患者在年轻时可能没有任何症状,但随着年龄增长,囊肿会不断增大增多,最终导致肾功能衰竭。
有研究显示,约25%的多囊肾患者会在47岁时发展到终末期肾脏疾病,50%在59岁时需要透析或肾移植,75%在70岁时会面临同样的境遇。
课程一览
1. 常染色体显性多囊肾病(ADPKD)与隐性多囊肾病(ARPKD)的区别
2. 基因突变与囊肿形成机制
3. 疾病进展自然史与长期预后
一、常染色体显性多囊肾病与隐性多囊肾病的区别
多囊肾病按照遗传方式可以分为两大类:常染色体显性多囊肾病和常染色体隐性多囊肾病。
这两种疾病虽然都叫多囊肾,但在发病年龄、严重程度、遗传特点等方面有着显著的区别。
1、发病年龄和临床表现的差异
常染色体显性多囊肾病,简称ADPKD,是成人型多囊肾,占所有多囊肾病例的90%以上。
这种类型的多囊肾有个特点,就是"姗姗来迟"。
大部分患者在儿童期和青少年期都没有明显症状,通常要到30-40岁以后才开始出现腰痛、血尿、高血压等症状。
有些"幸运"的患者甚至到了50-60岁才被发现。
这就像是一颗定时炸弹,平时看起来风平浪静,但实际上囊肿在悄悄地生长。
很多患者都是在体检时意外发现的,或者因为家族中有人患病而主动筛查才发现的。
常染色体隐性多囊肾病,简称ARPKD,则是幼儿型多囊肾,相对比较少见,发病率约为1/10000到1/40000。
这种类型的多囊肾就比较"急性子"了,通常在婴儿期或儿童期就会发病,而且症状往往比较严重。
2、遗传特点的区别
从遗传学角度来看,这两种疾病的遗传方式完全不同。
ADPKD是显性遗传,意思是只要从父母任何一方得到一个"坏"基因,就会发病。
如果父母中有一方患有ADPKD,那么每个孩子都有50%的概率会遗传这个疾病。
这就像是抛硬币一样,正反面各有一半的可能性。
ARPKD是隐性遗传,需要从父母双方各得到一个"坏"基因才会发病。
如果父母都是携带者(各有一个正常基因和一个异常基因),那么每个孩子有25%的概率会发病,50%的概率成为携带者,25%的概率完全正常。
3、疾病严重程度和预后差异
从疾病的严重程度来看,ARPKD通常比ADPKD更严重。
ARPKD患者在婴儿期就可能出现肾功能不全,需要早期干预治疗。
而且,ARPKD不仅影响肾脏,还常常伴有肝脏纤维化,形成所谓的"肝肾综合征"。
ADPKD虽然发病较晚,但疾病的进展相对缓慢。
大多数患者可以正常生活工作几十年,有些患者甚至终生都不会发展到需要透析的程度。
当然,这也因人而异,取决于基因突变的类型和个体差异。
4、肾外表现的特点
两种类型的多囊肾在肾外表现方面也有所不同。
ADPKD患者除了肾脏囊肿外,还可能出现肝囊肿、胰腺囊肿、脾囊肿等。
大约70-80%的ADPKD患者会出现肝囊肿,但通常不会影响肝脏功能。
更值得注意的是,ADPKD患者还可能出现脑动脉瘤,发生率约为8-10%,这是一个需要密切关注的并发症。
虽然大多数脑动脉瘤不会破裂,但一旦破裂就可能危及生命。
ARPKD患者的肾外表现主要是肝脏纤维化,这可能导致门静脉高压、食管静