课程一览
1. PKD1与PKD2基因功能异常
2. 肾小管上皮细胞增殖与囊肿形成
3. 纤维化进展与肾功能下降机制
4. cAMP信号通路与囊肿生长
一、PKD1与PKD2基因功能异常导致疾病发生
想要理解多囊肾,我们得先从基因说起。
就像每个人都有一套独特的DNA密码,我们的肾脏发育和功能也受到特定基因的调控。
在多囊肾患者中,主要是两个基因出了问题:PKD1和PKD2。
1、PKD1基因的关键作用
PKD1基因定位于染色体16p13.3,基因长度为47 kb,含有46个外显子,编码的蛋白质产物称为多囊蛋白1。
这个基因就像是肾脏细胞的"管理员",负责维持细胞之间的正常交流。
当PKD1基因发生突变时,这个管理员就"失职"了,细胞之间的沟通出现问题,正常的肾脏结构开始崩坏。
约80%~85%的患者是由PKD1基因突变引起的,这类患者通常症状比较严重,发病年龄也相对较早。
有意思的是,PKD1基因突变就像是一个"急性子",总是迫不及待地要表现出来,让患者在相对年轻的时候就开始出现症状。
2、PKD2基因的独特特征
相比之下,PKD2基因就"温和"一些。
PKD2基因定位于染色体4q21-23,基因长度为70 kb,含有15个外显子,编码的蛋白质产物称为多囊蛋白2,是一种通道蛋白。
约10%~15%的患者是由PKD2基因突变引起的。
具有PKD2基因突变的人群,常见于女性,与PKD1基因突变患者相比,通常患病程度较轻,其症状如肾功能下降等往往在成年后出现。
如果说PKD1是个"急性子",那PKD2就是个"慢性子",它让疾病的进展相对缓慢,给患者更多的时间来应对。
3、基因突变的遗传规律
多囊肾的遗传特点相当特殊。
只要父母双方中有一方具有致病的异常基因,孩子就会有50%的几率患上同样的疾病。
这就像是一个不公平的"抽奖",每个孩子都有一半的概率"中奖",而这个奖品却是疾病。
这种遗传方式叫做常染色体显性遗传,意思是只要有一个异常基因就会发病。
不像有些遗传病需要父母双方都有异常基因,多囊肾只需要一方有问题就够了。
这也解释了为什么很多多囊肾家庭会出现连续几代都有患者的情况。
二、肾小管上皮细胞增殖与囊肿形成的过程
了解了基因问题,我们再来看看这些基因异常是如何一步步导致囊肿形成的。
这个过程就像是一场连锁反应,一环扣一环,最终导致了肾脏结构的彻底改变。
1、正常肾小管的工作原理
在正常情况下,肾小管就像是一个精密的过滤和回收系统。
肾小管上皮细胞紧密排列,形成一个个小管道,血液经过这些管道时,废物被过滤出去,有用的物质被重新吸收回血液中。
这个过程需要上皮细胞之间的精密协调,就像一个训练有素的团队。
2、异常增殖的开始
当PKD1或PKD2基因异常时,肾小管上皮细胞就开始"不听话"了。
多囊蛋白1可能调节肾小管上皮细胞的黏附和分化;多囊蛋白2可能发挥离子通道的作用。
当这些蛋白功能异常时,细胞开始无序增殖,就像是失去了"刹车"的汽车。
正常情况下,细胞增殖是有严格控制的,只有在需要修复损伤或正常更新时才会分裂。
但在多囊肾患者中,这种控制机制失效了,细胞开始疯狂增殖,形成了异常的细胞团块。
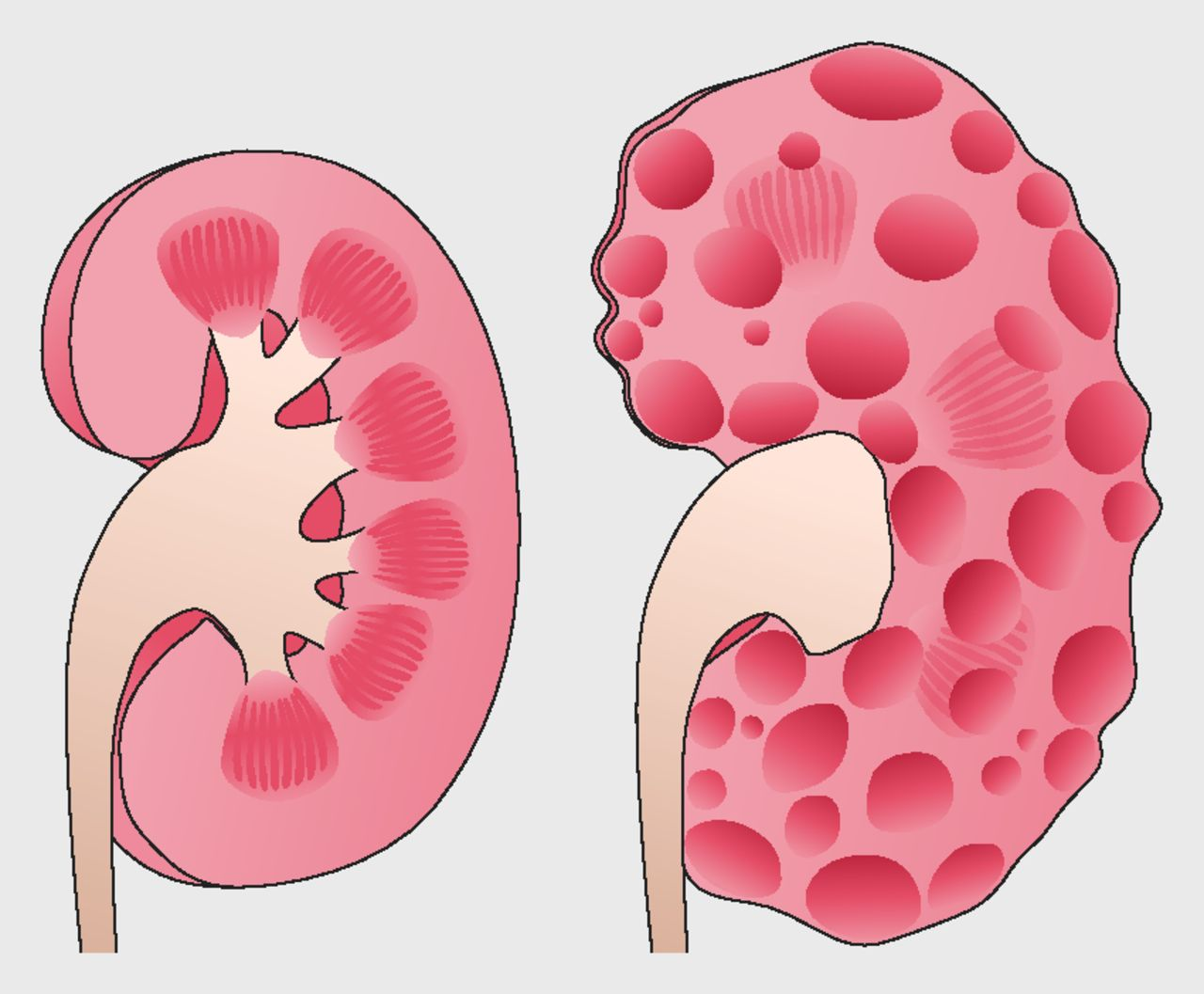
3、囊肿的逐步形成
细胞异常增殖只是第一步,接下来更关键的是囊肿的形成。
想象一下,原本整齐的肾小管突然出现了一个"鼓包",这个鼓包逐渐扩大,最终形成了一个充满液体的囊泡。
这些蛋白的突变可改变肾脏纤维化进程,其突变可导致液体向囊肿腔内分泌。
囊肿一旦形成,就会不断地吸引液体进入,就像一个永远填不满的水桶。
随着时间推移,这些囊肿会越来越大,数量也越来越多。
4、囊肿扩张的恶性循环
囊肿的扩张会进一步压迫周围的正常肾脏组织,造成更多的损伤。
这就形成了一个恶性循环:囊肿压迫正常组织,导致更多细胞受损,受损细胞又可能形成新的囊肿。
整个过程就像是多米诺骨牌效应,一旦开始就很难停止。
三、纤维化进展与肾功能下降机制
囊肿的形成只是多囊肾